What is validation?
Validation is a process of establishing documentary evidence demonstrating that a procedure, process, or activity carried out in production or testing maintains the desired level of compliance at all stages. In Pharma Industry it is very important apart from final testing and compliance of product with standard that the process adapted to produce itself must assure that process will consistently produce the expected results. Here the desired results are established in terms of specifications for out come of the process. Qualification of systems and equipment is therefore a part of process of validation. It is a requirement of food and drug, pharmaceutical regulating agencies like FDA's good manufacturing practices guidelines. Since a wide variety of procedures, processes, and activities need to be validated, the field of validation is divided into a number of subsections including the following:
• Equipment validation
• Facilities validation
• HVAC system validation
• Cleaning validation
• Process Validation
• Analytical method validation
• Computer system validation
• Packaging validation
• Cold chain validation
Reasons for validation?
FDA, or any other food and drugs regulatory agency around the globe not only ask for a product that meets its specification but also require a process, procedures, intermediate stages of inspections, and testing adopted during manufacturing are designed such that when they are adopted they produce consistently similar, reproducible, desired results which meet the quality standard of product being manufactured, such procedures are developed through the process of validation. This is to maintain and assure a higher degree of quality of food and drug products. Validation is "Establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes.". A properly designed system will provide a high degree of assurance that every step, process, and change has been properly evaluated before its implementation. Testing a sample of a final product is not considered sufficient evidence that every product within a batch meets the required specification.
The Validation Master Plan
The Validation Master Plan is a document that describes how and when the validation program will be executed in a facility. Even though it is not mandatory, it is the document that outlines the principles involved in the qualification of a facility, defines the areas and systems to be validated and provides a written program for achieving and maintaining a qualified facility with validated processes. It is the foundation for the validation program and should include process validation, facility and utility qualification and validation, equipment qualification, cleaning and computer validation. The regulations also set out an expectation that the different parts of the production process are well defined and controlled, such that the results of that production will not substantially change over time.
The Validation Process:
The validation scope, boundaries and responsibilities for each process or groups of similar processes or similar equipment's must be documented and approved in a validation plan. These documents, terms and references for the protocol authors are for use in setting the scope of their protocols. It must be based on a Validation Risk Assessment (VRA) to ensure that the scope of validation being authorised is appropriate for the complexity and importance of the equipment or process under validation. Within the references given in the VP the protocol authors must ensure that all aspects of the process or equipment under qualification; that may affect the efficacy, quality and or records of the product are properly qualified. Qualification includes the following steps:
Design qualification (DQ) - Demonstrates that the proposed design (or the existing design for an off-the-shelf item) will satisfy all the requirements that are defined and detailed in the User Requirements Specification (URS). Satisfactory execution of the DQ is a mandatory requirement before construction (or procurement) of the new design can be authorised.
Installation qualification (IQ) – Demonstrates that the process or equipment meets all specifications, is installed correctly, and all required components and documentation needed for continued operation are installed and in place.
Operational qualification (OQ) – Demonstrates that all facets of the process or equipment are operating correctly.
Performance qualification (PQ) – Demonstrates that the process or equipment performs as intended in a consistent manner over time.
Component qualification (CQ) – is a relatively new term developed in 2005. This term refers to the manufacturing of auxiliary components to ensure that they are manufactured to the correct design criteria. This could include packaging components such as folding cartons, shipping cases, labels or even phase change material. All of these components must have some type of random inspection to ensure that the third party manufacturer's process is consistently producing components that are used in the world of GMP at drug or biologic manufacturer.
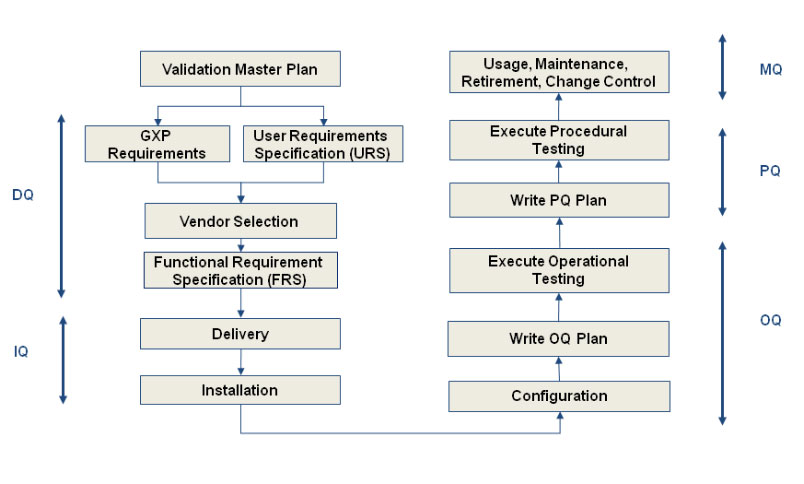
"Although PQ is described as a separate activity, it may in some cases be appropriate to perform it in conjunction with OQ.""Although PQ is described as a separate activity, it may in some cases be appropriate to perform it in conjunction with OQ."
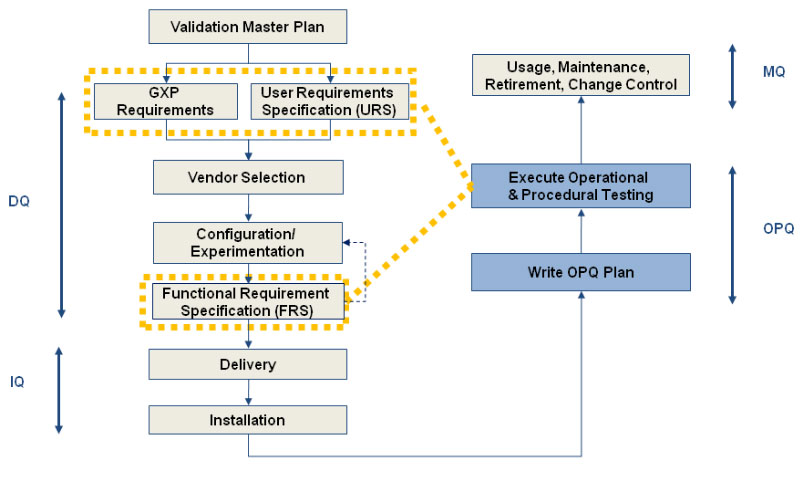
The Industry refers to Validation as “establishing documented evidence that a process or system when operated within the established parameters can perform effectively and reproducibly to produce a product which meets”.